

In this quick and easy guide, learn about Gastrointestinal System: Pathophysiology of Diseases and Cancers [Biology, MCAT, USMLE]. Topics include hepatobiliary disease, understanding liver functions and cirrhosis, liver function tests, causes of liver injury, alcoholic hepatitis, NAFLD, NASH, viral hepatitis, autoimmune hepatitis, drug-induced liver injury, hereditary hemochromatosis, Wilson’s disease, Alpha-1 Antitrypsin Deficiency, PSC, PBC, cholelithiasis, cholecystitis, cholangitis, hepatocellular carcinoma, Crohn’s disease vs Ulcerative Colitis, and more.
Table of Contents
Hepatobiliary Disease
Understanding Liver Functions and Cirrhosis, Broadly
Understanding “Liver Function Tests”
Causes of Liver Injury
Alcoholic Hepatitis
NAFLD/NASH
Viral Hepatitis
Autoimmune Hepatitis
Drug-Induced Liver Injury
Hereditary Hemochromatosis
Wilson’s Disease (Hepatolenticular Degeneration)
Alpha-1 Antitrypsin Deficiency
Primary Sclerosing Cholangitis
Primary Biliary Cirrhosis
The “chole” friends
Cholelithiasis
Cholecystitis
Choledocholithiasis
Cholangitis
Neoplasms
Hepatocellular Carcinoma
Hepatic adenomas
Focal nodular hyperplasia
Esophagogastric Disease
Intestinal Disease
Inflammatory Bowel Disease
Crohn Disease
Ulcerative Colitis
Diverticulosis vs Diverticulitis
Neoplasms
Polyps (including polyposis syndromes)
Pancreatic Disease
Hepatobiliary Disease
Understanding Liver Functions…and what happens when they’re gone = cirrhosis / end-stage liver disease
To understand cirrhosis – i.e., what happens when the liver stops working, you need to understand the normal functions of the liver in health. Broadly, these can be divided into: (1) synthetic functions (proteins, lipids) // (2) detoxification (drugs, toxins, metabolic wastes, especially ammonia) // (3) bile production and excretion // (4) carbohydrate metabolism (e.g., gluconeogenesis, glycogen synthesis & lysis) // (5) lipid metabolism // (6) micronutrient storage (fat-soluble vitamins = ADEK + B12, Fe). I will discuss a few of these & how their loss presents in patients with end stage liver disease (i.e., cirrhosis):
Synthetic functions
- Hepatocytes are the only cells that synthesize:
- Albumin (main protein responsible for maintaining oncotic pressure in blood)
- Fibrinogen (clotting factor I)
- Prothrombin group of clotting factors (X / IX / VII / II; “1972” + proteins C and S) — notably, these are also the Vitamin K-dependent clotting factors (i.e., they require Vitamin K to be activated via gamma-carboxylation, and are the target of the anticoagulant warfarin, which is a Vitamin K epoxide reductase inhibitor).
- They otherwise are the most significant source of:
- Lipoproteins (LDL, VLDL, HDL, etc.)
- I had a patient with cirrhosis with an HDL <5. It wasn’t because he didn’t eat enough almonds, it was because his hepatocytes couldn’t make apolipoproteins C and E.
- Ceruloplasmin (carries Cu) — more to come with Wilson’s Disease below, yay!
- Transferrin (carries Fe)
- Glycoproteins
- Lipoproteins (LDL, VLDL, HDL, etc.)
- This is clinically relevant in that defects in hepatic synthetic function are reflected clinically by:
- Decrease in albumin (hypoalbuminemia)
- Increase in prothrombin time/INR
- Just to reinforce/expand a bit since y’all haven’t done heme – prothrombin time (PT), is a measure of the extrinsic + common pathways for clotting. Basically, to perform the test you add Ca2+ and thromboplastin (an activator of the extrinsic pathway) to a patient’s blood, then see how long it takes for the patient’s blood to clot (i.e., generate fibrin cross links).
- INR stands for International Normalized Ratio and is a measure that allows for inter-institution comparison of prothrombin times, and is just a ratio of the observed prothrombin time to a “standard” PT. On the wards, you’ll generally report both PT and INR, but INR is technically more accurate.
- Take home point: longer INR = longer time to clot = more tendency to bleed. Normal is 0.9-1.3.
- Moreover, the extent of prolongation of the INR is a valuable prognostic factor for predicting mortality in end-stage liver disease (see MELD Score / Child Pugh classification below).
Metabolism / detoxification of endogenous hormones, drugs, and toxins
- As you may recall from pharmacology, hepatic drug metabolism occurs in two phases, conveniently named Phase I and Phase II.
- In Phase I, lipophilic compounds are made more water-soluble via addition of polar groups via redox and hydrolysis reactions.
- These reactions are catalyzed by the cytochrome P450 system (e.g., CYP2E1, CYP3A4).
- Side note: there’s a whole list of cytochrome P450 inducers and inhibitors at the end of the pharm section of First Aid (p243 from the 2017 edition).
- Don’t worry about memorizing them just now, but do:
- (1) know that this list is there and is VERY HIGH YIELD
- (2) go ahead and acquaint yourself with the idea that a cytochrome INHIBITOR will lead to decreased metabolism / increased drug effect and toxicity, while an INDUCER will lead to increased metabolism / decreased drug effect and toxicity.
- You will almost ~certainly~ have a question about this on Step 1, likely dealing with how warfarin’s effect on INR varies with co-administration of other drugs (e.g., prolonged INR with an inhibitor like ciprofloxacin, metronidazole, or amiodarone).
- Another classic = acetaminophen and EtOH…
- These reactions are catalyzed by the cytochrome P450 system (e.g., CYP2E1, CYP3A4).

- Note, in the above, that NAPQI (the hepatotoxic metabolite) is generated in a reaction catalyzed by CYP2E1. Therefore, CYP2E1 inhibitors will decrease generation of this toxic metabolite, while inducers will increase its generation. This leads to a surprising observation regarding the differential effects of acute vs chronic EtOH on acetaminophen toxicity:
- Acutely, EtOH is a CYP inhibitor –> dec. NAPQI –> less acetaminophen toxicity noted with acute alcohol intoxication.
- Chronically, EtOH is a CYP inducer –> inc. NAPQI –> more acetaminophen toxicity noted in patients with chronic alcohol over/misuse. Patients who overuse EtOH may also of course have baseline liver dysfunction (see section on alcoholic hepatitis below), which would also predispose them to a worse outcome in s/o acetaminophen overdose.
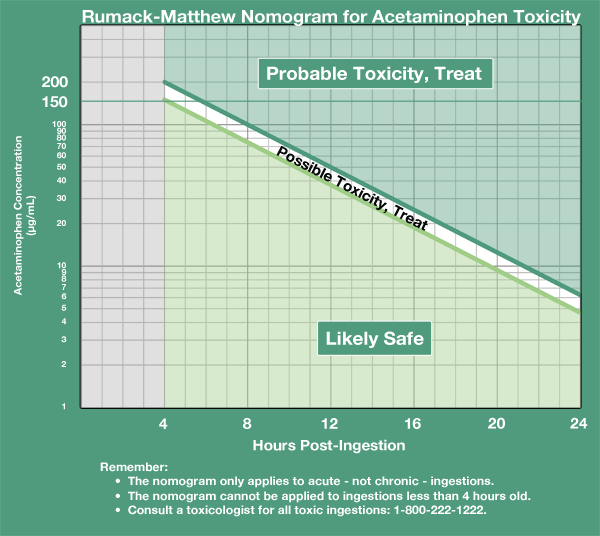
- And just while we’re on the topic, recall that the antidote for acetaminophen toxicity is N-acetylcysteine, given in accordance with the Rumack-Matthew nomogram (which gives the need for NAC in terms of current acetaminophen level and hours since acetaminophen ingestion).
- Phase II involves conjugation reactions, i.e., addition of highly water soluble groups
- The mnemonic is GAS: glucuronidation, acetylation, and sulfation
- Glucuronidation is one of the steps in bilirubin metabolism (see below).
- These reactions are great because highly polar/charged molecules cannot cross biological membranes to have a toxic effect, and will be peed out instead.
- Clinical tie-in: conjugated bilirubin cannot cross the blood-brain barrier to cause kernicterus (or bilirubin-induced neurologic dysfunction, as the cool kids are calling it these days), as bilirubin glucuronide is super mega polar (look at all those -OH and COO- groups!!!)
- This is NOT to say that a conjugated hyperbilirubinemia (again, for more discussion/explanation, see the below section on bilirubin metabolism) in infancy is not dangerous / should not be treated, but rather to reinforce the idea that (1) glucuronidation is the natural mechanism for
- Very polar/charged – highly water-soluble – cannot cross biological membranes to have a toxic effect – will be peed out instead
- What might deconjugate xenobiotics? Why is this significant?
- Colonic bacteria – can have toxic effect again
- Okay, but what if I have cirrhosis and the above reactions aren’t working?
- You get increased drug toxicity from drugs with a primarily hepatic metabolism. The list of drugs that are metabolized hepatically is impossibly long to memorize, but just know to be aware of this and to look up drug adjustments for hepatic failure using Epocrates, Lexicomp, UpToDate, or another resource when you have a patient with end-stage liver disease.
I’m making ammonia detoxification its own section b/c it’s such a huge piece to understand. Recall (and so begins the secondary biochem retraumatization, bear with me and my undergrad degree of choice) that the liver is the site of the urea cycle…
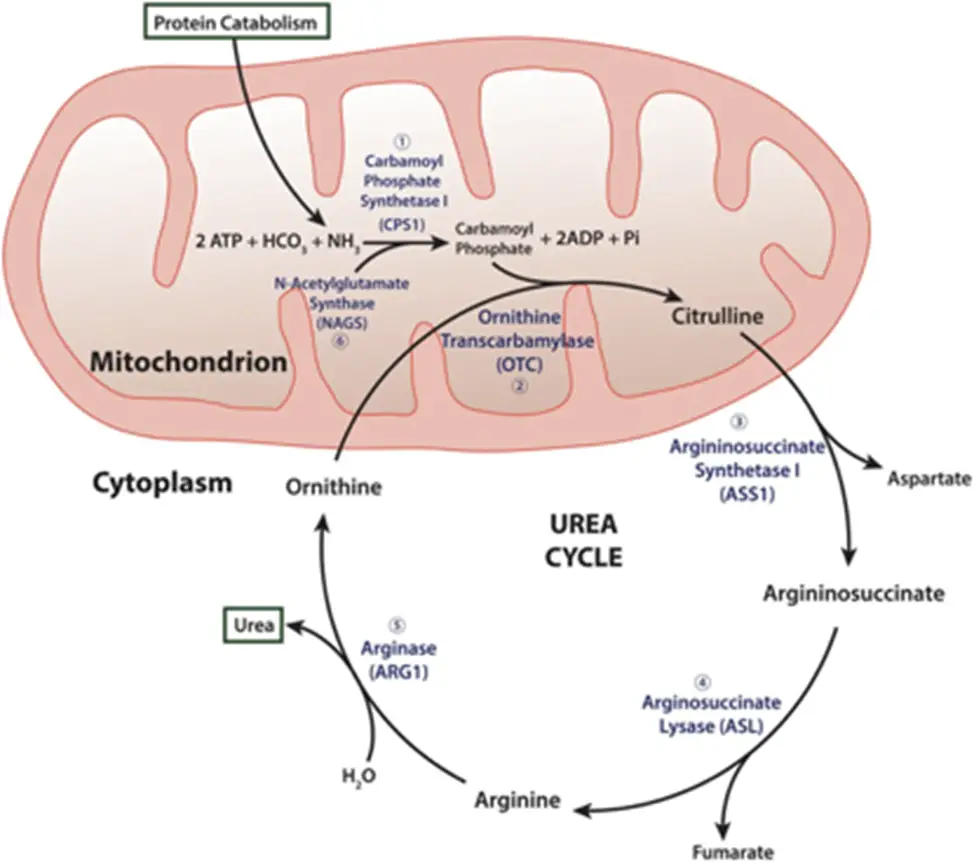
- Remember me?? Good ol’ “ordinarily, careless crappers are also frivolous about urination?” DO NOT WORRY, you absolutely do not need to memorize these specifics for this exam (Step 1 being a different story…); I’m just including this here to increase the salience of the idea that the liver is the site of ammonia detoxification. That is, the “point” of the urea cycle is to convert excess nitrogen from the body’s proteins and nucleic acids into a non-toxic form that can be easily excreted in the urine – i.e., urea!
- Ammonia (NH3) is toxic predominantly to the CNS. How? As usual, the exact mechanism is debated, but the following likely contribute (remember, if it’s “debated”, it’s low yield…):

2. The above is what happens ACUTELY, but over time, glutamate (NMDA) receptors are down-regulated due to excess glutamatergic tone and GABA-ergic tone predominates (GABA, or gamma-aminobutyric acid, is the main inhibitory neurotransmitter in the CNS)
a. This can help you understand the clinical manifestations of hepatic encephalopathy, the state of altered mental status that accompanies liver dysfunction. In general, patients will become less responsive / more confused/obtunded, begin sleeping during the day instead of the night, and progress gradually to a coma. There is a characteristic hand tremor known as asterixis, which occurs when patients attempt to maintain a position of hands extended with outstretched arms (easier to witness than explain, so take a look).
i. Note: while asterixis is characteristic of HE, it is not specific for it – other metabolic encephalopathies, including uremic and hypercapnic encephalopathy, can cause patients to have a similar tremor.
ii. There are 4 stages of HE, included here for reference, probably more high yield for the wards than this exam.
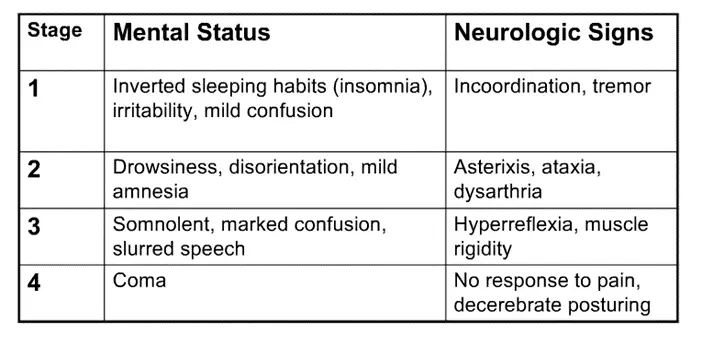
- Ataxia = incoordination, generally due to cerebellar dysfunction (as cerebellum helps coordinate movements)
- Dysarthria = slurred speech
- Decerebrate posturing = abnormal extension of arms / legs due to disconnection of motor neurons from normal input from CNS (due to the encephalopathy); you’ll learn more specifics on neuro
- The mental status of a patient HE (or hyperammonemia from any cause, such as urea cycle defects or organic acidemias) can therefore be improved by administering drugs that increase NH3 excretion. These include: lactulose and rifaximin (there are others under study, but you for SURE need to know these).
- Lactulose – carbohydrate that’s chewed up by colonic flora, which produce acid, thereby protonating NH3 in the gut and trapping it as NH4+ for excretion

- Note – lactulose is also just a really good laxative, and there’s a theory that the acid trap hypothesis is kind of hogwash and it’s just the overall loss of nitrogenous waste (i.e., proteins and nucleic acids in foods) that contributes to the improvement in MS.
- This also contributes to noncompliance – would you want to take a drug 3-4 times per day that had an excessively sweet taste and caused bloating, flatulence, and severe and unpredictable diarrhea? Me either…
- Rifaximin – PO antibiotic that’s minimally absorbed (i.e., minimal side effects) that gets concentrated in the GI tract and can kill off NH3-producing colonic flora. It works super well and doesn’t seem to contribute to antibiotic resistance, so, hell yes.
- In addition, it’s important to correct hypokalemia if present, since hypokalemia increases renal ammoniagenesis (don’t worry about mechanism).
- HOWEVER: if someone asks you on Internal Medicine (hint: this will likely happen) whether or not trending ammonia levels is helpful in managing a patient with hepatic encephalopathy, the answer is NO – hyperammonemia is central to the pathogenesis of HE, but serial ammonia levels don’t inform a diagnosis of HE – it’s a clinical diagnosis. That is, a patient can be diagnosed with HE with a normal ammonia level, AND patients with abnormal ammonia levels can have a normal mental status, so the test is neither sensitive nor specific.
- That said, a markedly elevated ammonia level (e.g., >100) can carry prognostic significance in acute liver failure (or other causes of hyperammoemia), so it’s not a USELESS test, but just make sure to interpret the result carefully.
- So — why do you get excess serum NH3 in cirrhosis anyway?
- Dysfunctional hepatocytes aren’t particularly good at the urea cycle
- When your liver fibroses, the nice spacious sinusoids are now full of scar tissue and you get elevated back pressures within the portal system (so-called portal hypertension). Porto-systemic collaterals (image and further explanation below) then develop, which allow toxins from the GI tract (i.e., NH3) to bypass the liver and enter the systemic circulation without first undergoing hepatic metabolism.
- Note also that when you have really severe portal hypertension (e.g., refractory variceal bleeds, horrible ascites), you may need a procedure like a TIPS (transjugular intrahepatic porto-systemic shunt), which creates a connection between the portal vein and the hepatic veins to relieve portal HTN. While this may improve the sequelae of portal HTN, a possible side effect will be worsening of hepatic encephalopathy.

- Let’s take this one step further – when a patient with cirrhosis eats a big protein load, the nitrogenous burden of this meal may overwhelm the liver’s detoxifying capacity, and HE may result. Other precipitants of hepatic encephalopathy include:
-
- GI bleed (nitrogenous burden of blood proteins)
- Renal failure (azotemia = inc nitrogen in blood)
- Constipation (more time to absorb nitrogenous wastes and yet another reason to give lactulose)
- CNS depressants (e.g., benzos) – no surprise that they could worsen mental status…
- Diuretic-induced hypovolemia (recall that you can get a contraction alkalosis from overly aggressive diuresis [multifactorial but contributors are likely increased AII = more Na/H exchange in PCT = more proximal reabsorption of bicarb and more aldo = more H+ excretion in DCT]; the alkalosis will worsen hyperammonemia b/c while NH4+ is ‘trapped’ in the lumen and will be excreted in the urine, NH3 can be reabsorbed into the bloodstream
- And recall that both loop and thiazide diuretics cause hypokalemia, which as noted above, increases renal ammoniagenesis
- Why should you care about HE precipitants? b/c you need to know whether there’s something reversible to help your patient with HE get better!
-
Production of bile, including conjugation and excretion of bilirubin
OK — bilirubin metabolism can seem a bit hairy, but a review of some core concepts should help make things clear, and I’ll tie in relevant diseases as they come up:
- Breakdown of heme (the Fe-containing, O2-transporting coordination complex at the center of hemoglobin in RBCs) from old RBCs via phagocytes (macrophages) in the spleen, liver, and bonemarrow creates bilirubin.
- There’s a bit more detail here, as this occurs in 2 reactions:
- Heme –> biliverdin via heme oxygenase (requires oxygen, as hinted in the name)
- This is the rate-limiting step in bilirubin production.
- Biliverdin –> bilirubin via biliverdin reductase (requires NADPH, like most biological reductions)
- ^^both of these reactions occur within the macrophage.
- Bilirubin then binds very avidly to albumin within the serum.
- Heme –> biliverdin via heme oxygenase (requires oxygen, as hinted in the name)
- A key clinical tie-in here is that diseases that cause increased RBC destruction, i.e., hemolysis, will cause an unconjugated hyperbilirubinemia (more on conjugation below). Hemolysis will be covered in much more detail on heme, but here’s a brief outline of some high-yield hemolytic diseases:
- Hereditary spherocytosis (#TBT CFM!!) – results from mutations in RBC membrane-anchoring proteins (spectrin, ankyrin, band 3, protein 4.2), RBC membrane is more labile to losing little bits of its membrane in tiny vesicles; as the surface area-to-volume ratio of the RBC decreases, the cell adopts a more spherical (rather than biconcave) shape. Since a sphere is less deformable than the disc, as the RBCs get filtered in the spleen (specifically as they move through the cords of Bilroth into the sinusoids), they get stuck and chewed up by splenic macrophages, causing hemolysis. Patients will therefore present with anemia (easy fatigability, pallor, tachycardia, systolic flow murmur over aortic valve due to hyperdynamic circulation to meet O2 demands of tissues despite loss in O2 carrying capacity), jaundice, splenomegaly, and complications of pigmented gallstones (e.g., biliary colic, cholecystitis, cholangitis – more on these below). HS is inherited in an autosomal dominant manner and is most common in patients of Northern European descent.
- Glucose 6 Phosphate Dehydrogenase deficiency – just when you thought you were done with the pentose phosphate pathway forever!!! Let’s break this down…
- RBCs use glutathione as the primary antioxidant to reduce dangerous ROS (reactive oxygen species) into harmless alcohols/water.
- In this process, “-GSH” (reduced glutathione) becomes “GS-SG” (oxidized glutathione).
- You need NADPH to regenerate GSH from GS-SG.
- Where do you get NADPH? From the first (oxidative) step of the pentose phosphate pathway, which takes G-6-P to R-5-P via glucose 6 phosphate DH, consuming NADP+ and generating NADPH.
- RBCs use glutathione as the primary antioxidant to reduce dangerous ROS (reactive oxygen species) into harmless alcohols/water.
- There’s a bit more detail here, as this occurs in 2 reactions:
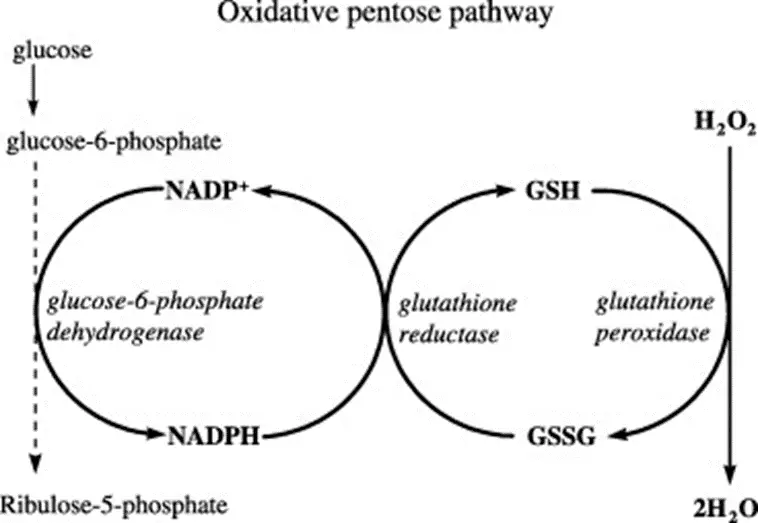
- Therefore, if you have a deficiency in G6PD, you will be unable to generatenew NADPH within RBCs, which will thereby be exposed to more oxidative stress. When the degree of oxidative stress exceeds the reductive capacity of the RBC, hemolysis results.
- This disorder is X-linked recessive (boys, boys! + girls with unfavorable lyonization…) and most common in patients of Mediterranean descent. You may recall the association with fava beans, which contain several oxidizing substances and can precipitate hemolytic crises in patients with G6PD deficiency. Certain drugs may do this as well, including sulfonamides (e.g., TMP/SMX), dapsone, anti-malarials (e.g., primaquine, hydroxychloroquine, pyrimethamine), INH, ibuprofen, ASA, and nitrofurantoin.
- Sickle Cell Disease
- Microangiopathic hemolytic anemia – TTP, HUS, DIC – refer to the renal doc for a review of these
- Autoimmune Hemolytic Anemia
- So what? Why should we care about elevated bilirubin?
- Bilirubin is neurotoxic (our favorite!! #TBTNH3) in neonates, who have an immature blood-brain barrier.
- Specifically, bilirubin can deposit within the deposit within basal ganglia, a condition known as kernicterus or bilirubin-induced neurologic dysfunction (BIND)
- Acutely, severe neonatal hyperbilirubinemia presents with lethargy, poor feeding, increased or decreased muscle tone, seizures, and possibly death. If the serum bilirubin level is not rapidly lowered via exchange transfusion (taking patient’s blood and replacing it with normal blood) – and potentially even if it is – the child will go on to have chronic bilirubin encephalopathy, characterized by movement disorders (unsurprising given deposition in basal ganglia; see above comment), hearing loss, decreased eye movement, and GI motility issues.
- This is a pediatrician’s (and, obviously, parent’s) worst nightmare, and is why bilirubin is monitored serially in the hours to days after birth to ensure levels never approach kernicterus-inducing levels; more on the measurement and management of neonatal hyperbilirubinemia below once we cover more of the basic science.
- Specifically, bilirubin can deposit within the deposit within basal ganglia, a condition known as kernicterus or bilirubin-induced neurologic dysfunction (BIND)
- After the neonatal period, hyperbilirubinemia generally does not cause neurologic dysfunction, but will present with jaundice (skin yellowing, particularly noticeable as scleral and sublingual icterus – eyes and tongue are super helpful places to look if your patient’s skin is dark); if the hyperbilirubinemia is due to an obstruction to bile flow, retention of bile salts will also cause intense pruritis.
- Bilirubin is neurotoxic (our favorite!! #TBTNH3) in neonates, who have an immature blood-brain barrier.
- Back to the basics: bilirubin cannot be filtered by the kidney (i.e., excreted in the urine) because it’s bound too tightly to albumin.
- What’s our solution? Conjugation, specifically via glucuronidation.
- Recall from above that this is a Phase [II] reaction, and like all reactions of this phase, it dramatically increases the water-solubility of bilirubin. CB can therefore dissociate from albumin and be filtered by the glomerulus
- Because of this, patients with conjugated hyperbilirubinemia will have associated bilirubinuria.
- Recall from above that this is a Phase [II] reaction, and like all reactions of this phase, it dramatically increases the water-solubility of bilirubin. CB can therefore dissociate from albumin and be filtered by the glomerulus
- What’s our solution? Conjugation, specifically via glucuronidation.
- How does this happen? albumin-bound UCB freely enters the Space of Disse through the fenestrated sinusoidal epithelium, then UCB alone dissociates from albumin and is taken up into the hepatocyte via OATP1B1, an organic anion transporter.
- It then goes to the SER for conjugation via UDP-glucuronyltransferase (fancy word, but just means it adds a highly water soluble glucuronyl group with the help of uridine diphosphate, the friendly RNA nucleotide).
- Why do we care about this enzyme? 2 diseases result if it’s messed up:
- Crigler-Najjar syndrome
- Type I – NO UDP-glucuronyltransferase activity – severe hyperbilirubinemia, early death + kernicterus
- Type II – moderately decreased UDP-glucuronyltransferase activity – less severe; will respond to phenobarbital (a CYP450 inducer).
- Gilbert syndrome
- Results from a mild decrease in UDP-glucuronyltransferase activity, with a generally mild, asymptomatic increase in serum bilirubin levels that occurs only with fasting, stress, or processes causing increased RBC breakdown.
- INTERESTINGLY, patients with Gilbert syndrome are actually at LOWER risk for coronary artery disease, a fact that’s attributed to the antioxidant properties of bilirubin. A little pigment never hurt nobody?
- As I’ll emphasize below when I discuss the ddx of conjugated vs unconjugated hyperbilirubinemia, both of these illnesses will result in an unconjugated hyperbilirubinemia…which is hopefully unsurprising given that they result from defects in…conjugating bilirubin.
- Crigler-Najjar syndrome
- Why do we care about this enzyme? 2 diseases result if it’s messed up:
- Conjugated bilirubin then heads into the bile canaliculi for excretion in an ATP-dependent mechanism via ABCC2 (ATP-binding cassette C2), also known as multidrug resistance protein 2 (MRP2).
- Why do we care about this little protein?
- Mutations/decreased function results in Dubin-Johnson syndrome, a disease in which there is a mild conjugated hyperbilirubinemia that, like Gilbert syndrome, is worse with intercurrent illness, fasting, stress, or hemolysis.
- Mutations in OATP1B1 (the organic anion transporter that lets bilirubin into the hepatocyte, see above) cause a very similar illness called Rotor syndrome.
- How can we distinguish between the two?
- In DJS – the bilirubin can get into the hepatocyte easily, so the liver will have a dark black pigment.
- Conversely, in RS, the liver is normally pigmented because the issue is with bilirubin uptake into the hepatocyte.
- Just to be super explicit: in contrast to Gilbert and Crigler-Najjar, which result defects in bilirubin conjugation and present with unconjugated hyperbilirubinemia, DJ causes a conjugated HB because the defect occurs after bilirubin is conjugated. RS also causes a conjugated HB, in this case because while bilirubin cannot get into the hepatocyte as easily, the bilirubin that DOES make it in can still be conjugated normally.
- Why do we care about this little protein?
- Here’s a summary of all that:
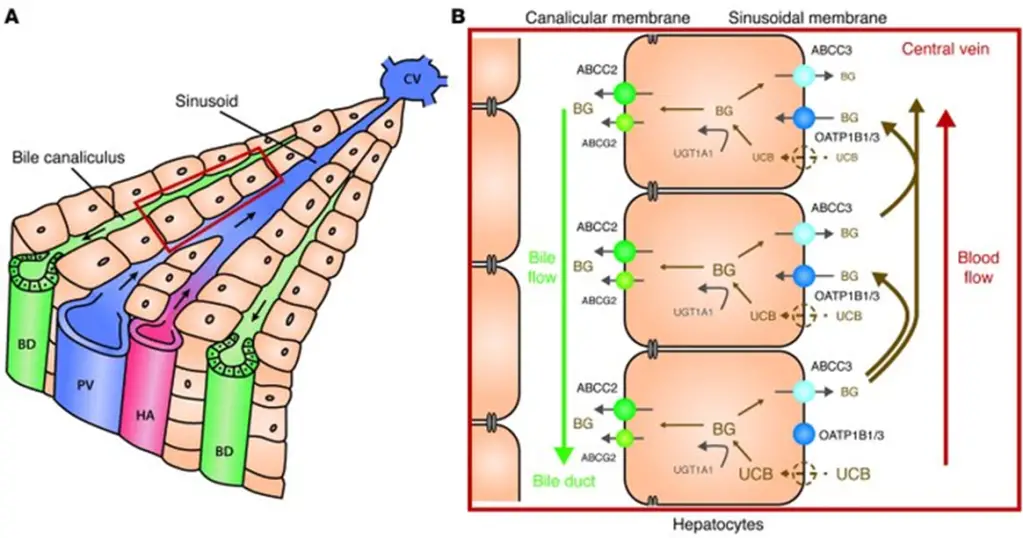
- As you look through the above image, review each disease:
- [Rotor syndrome] causes a [conjugated conjugated or unconjugated?] HB due to deficits in OATP1B1.
- [Gilbert syndrome] and [Crigler-Najjar syndrome] cause [unconjugated conjugated or unconjugated?] HB due to deficits in UGT1A1.
- [Dubin-Johnson syndrome] causes a [conjugated conjugated or unconjugated?] HB due to deficits in ABCC2/MRP2.
- OK – once bilirubin is in the bile canaliculus, it heads out of the liver with into the hepatic duct, where it then can either enter the gallbladder for storage via the cystic duct, or head into the intestine for excretion via the common bile duct, where it’ll join with the pancreatic duct to dump into the (2nd portion of the) duodenum at the ampulla of Vater.
- Imagine now if you had some obstruction to normal bile flow out of the liver,
- Oxidation of [UCB] by [colonic flora].
- In what age group is enterohepatic circulation significant?
- [Neonates] – because they lack [anaerobic gut flora until 6-8 wks].
Abnormal
- True or false: jaundice is generally noticeable prior to abnormalities in serum bilirubin.
- False; jaundice is not noticeable until a Tsb of 3.5.
- “Direct” bilirubin represents [conjugated] bilirubin.
- Any elevation above [15%] of total bilirubin represents some degree of [conjugated] hyperbilirubinemia; especially concerning when above [30%].
- In general, what causes…
- Conjugated HB?
- Diseases of the [biliary tract] or [hepatic parenchyma].
- Unconjugated HB?
- Diseases causing [hemolysis], alterations in [blood flow to liver], or [genetic disorders of conjugation].
- Which is usually more concerning – conjugated or unconjugated HB?
- Conjugated
- Conjugated HB?
Unconjugated HB
- #1 cause on your ddx: [hemolysis], including
- [G6PD deficiency]
- [Hereditary Spherocytosis]
- [Autoimmune hemolytic anemia]
- [Wilson’s disease]
- What diseases or states can cause decreased delivery of UCB to hepatocytes?
- [R heart failure]
- [porto-systemic shunts]
- Give some examples of diminished clearance or failure of storage of UCB
- Drug: [rifampicin]
- States with decreased ligandin: [pregnancy] + [hypothyroidism] + [febrile illness]
- What about failure of glucuronyl transferase enzymes?
- Mild: [Gilbert] – usually precipitated by [stress], [infections], or [fasting]
- Severe: [Crigler-Najjar] – Type I [deadly in infancy] without [liver transplantation], Type II [less severe]
- What contributes to neonatal HB?
- Immature [liver / conjugative enzymes]
- Increased [enterohepatic circulation] due to [insufficient flora] and [suboptimal feeding] in first days of life
- Elevated [hematocrit] at birth – higher [bilirubin] load
- [Prematurity]
- [Hemolysis] due to [ABO / Rh] incompatibility
- Why is neonatal HB dangerous?
- [UCB] can cross [blood-brain barrier] and
- How do we prevent this?
- [phototherapy] – uses [blue light] to create soluble isomer that does not need conjugation
- [frequent feeding] – increase intestinal transit speed / decrease [enterohepatic circulation]
- If severe/refractory – [exchange transfusion]
Conjugated HB
- Two broad causes:
- [impaired secretion into canaliculi]
- [obstruction to biliary flow]
- What is definitively diagnostic?
- [bilirubinuria]
- What happens to stool color, and why?
- [loss of bile pigments from stool – become ‘acholic’]
- Defects in MRP2 cause what disease?
- [Dubin Johnson] syndrome – can be distinguished from [Rotor] syndrome by the presence of [black pigment] in hepatocytes.
- The drug [cyclosporine] can also cause a similar defect.
- How can you distinguish CHB due to cholestasis from that due to hepatocellular disease?
- Cholestasis – markedly elevated [alk phos] – [3] times ULN – and [cholesterol], [AST / ALT] usually closer to normal, may complain of [pruritis] and have [steatorrhea], if [INR] prolonged, will respond to [Vitamin K]
- What classically causes a mixed pattern of injury?
- [alcoholic hepatitis] + [drug-induced liver injury, DILI]
- What besides cholestasis will increase alk phos?
- [puberty]
- [blastic bone mets]
- [renal disease]
- [pregnancy]
- What test can you send to distinguish between hepatocellular vs bone origin of Alk phos? –> [GGT]
- Cholestatic workup
- Imaging: [RUQ U/S] is best first test, then [CT abdomen] if nondiagnostic
- If suspect intrahepatic cholestasis –> [liver bx]
- If suspect extrahepatic obstruction / imaging w/ [dilated ducts] –> [ERCP]
- [EUS] is adjunctive to look at pancreatic head tumors / potentially bx
Maintenance of low portal pressures
- Portal hypertension, porto-caval anastomoses, ascites (don’t forget SAAG)

- So to summarize, let’s explore several of the clinical manifestation of cirrhosis and attempt to match them with some combination of the above
I’ve mentioned several of the clinical manifestations of cirrhosis in bits and spurts above, but I think it’ll be helpful for me to reiterate them here with tie-ins to the underlying pathophys to help bring everything together (and move away from the biochem trees to the overall forest).
First of all, know that pathologically, cirrhosis is defined by
- scites – multifactorial in etiology (classic), but pathophysiologic contributors include:
- Increased NO (for unclear reasons) leading to splanchnic vasodilation (the “under-filling” hypothesis you may recall from renal)
- Gynecomastia
- Hormonal production is disordered (again, who knows exactly why), with inc androstenedione from adrenals
- Spider angiomata / palmar erythema
- Asterisks
- Hepatic Encephalopathy
- Testicular atrophy / hypogonadism
- Purpura – synthetic dysfunction (coagulopathy)
- Edema
- Caput medusae – portal HTN
- MELD
- Child Pugh
- Complications of cirrhosis – if a patient has any of these, he/she has “decompensated” cirrhosis
- Variceal hemorrhage
- Ascites
- Spontaneous bacterial peritonitis
- Hepatic encephalopathy
- Hepatocellular carcinoma
- Hepatorenal syndrome
- Hepatopulmonary syndrome
Liver Function Tests?
Before we delve into individual diseases that can cause liver injury, let’s deconstruct what people mean when they say “LFTs” (liver function tests) – a non-specific and sometimes unhelpful catchall for several independent tests, each of which can be elevated OR normal in hepatic and non-hepatic disease. Clear as mud? Let’s take them one by one:
- AST/ALT
- Alkaline phosphatase
- Gamma-glutamyl transferase
- Bilirubin
- Unconjugated (Indirect)
- Conjugated (Direct)
- Albumin
Now that you understand a bit about each of those, let’s divide patterns of injury into 2 broad categories that are useful for the ddx of hepatic injury:
- Hepatocellular pattern of injury
- Cholestatic pattern of injury
Causes of Liver Injury
**to begin, a caveat — many of these etiologies can cause acute and chronic liver failure, depending on the timing of the insult. Infection with HBV, for instance, can cause an acute fulminant hepatic failure and can go on to cause chronic hepatitis contributing to the development of cirrhosis and hepatocellular carcinoma. Alcoholic hepatitis is similar. I will try to make these distinctions clear in the sections that follow, but just keep that in mind.
Zones of Liver Injury
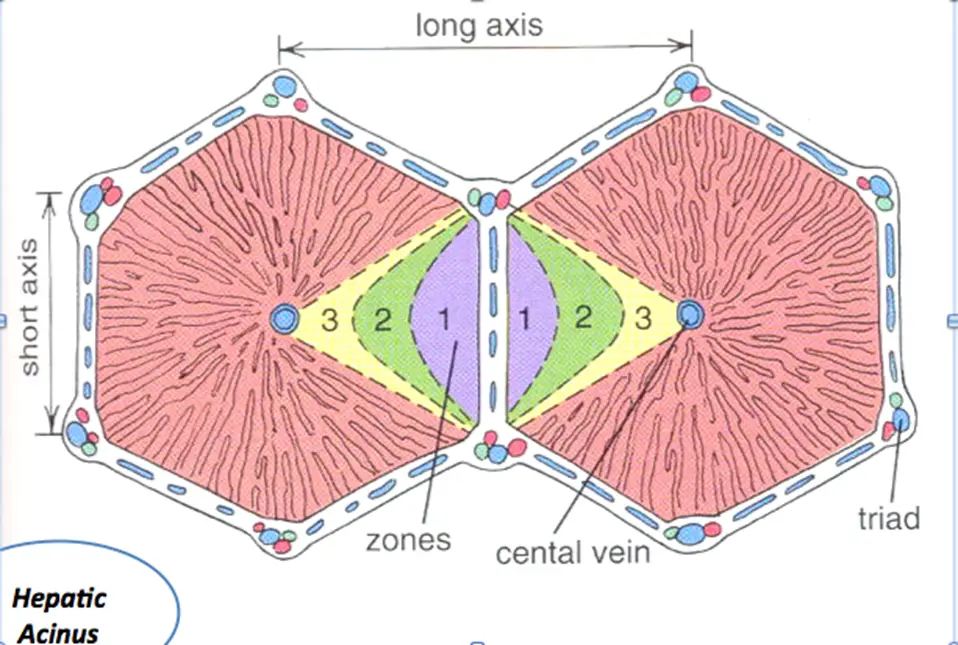
- Zone 1 – closest to blood supply – therefore most affected by viral hepatitis, cocaine, bile duct blockage (cholestasis)
- Zone 2 – most affected by Yellow fever (idk, yo)
- Zone 3 – farthest from blood supply and has P450 system that metabolizes drugs/toxins – therefore predisposed to ischemia, alcoholic hepatitis, and metabolic toxins
Alcoholic Hepatitis
- Pathogenesis / Etiology / Pathophysiology
- OK – if you’ve made it this far without knowing that alcohol is bad for your liver, then I’m not quite sure what to say. HOWEVER, there are some ~fun biochem reasons~ to know exactly why (which are more relevant for Step 1 than this exam, but I can’t help myself)

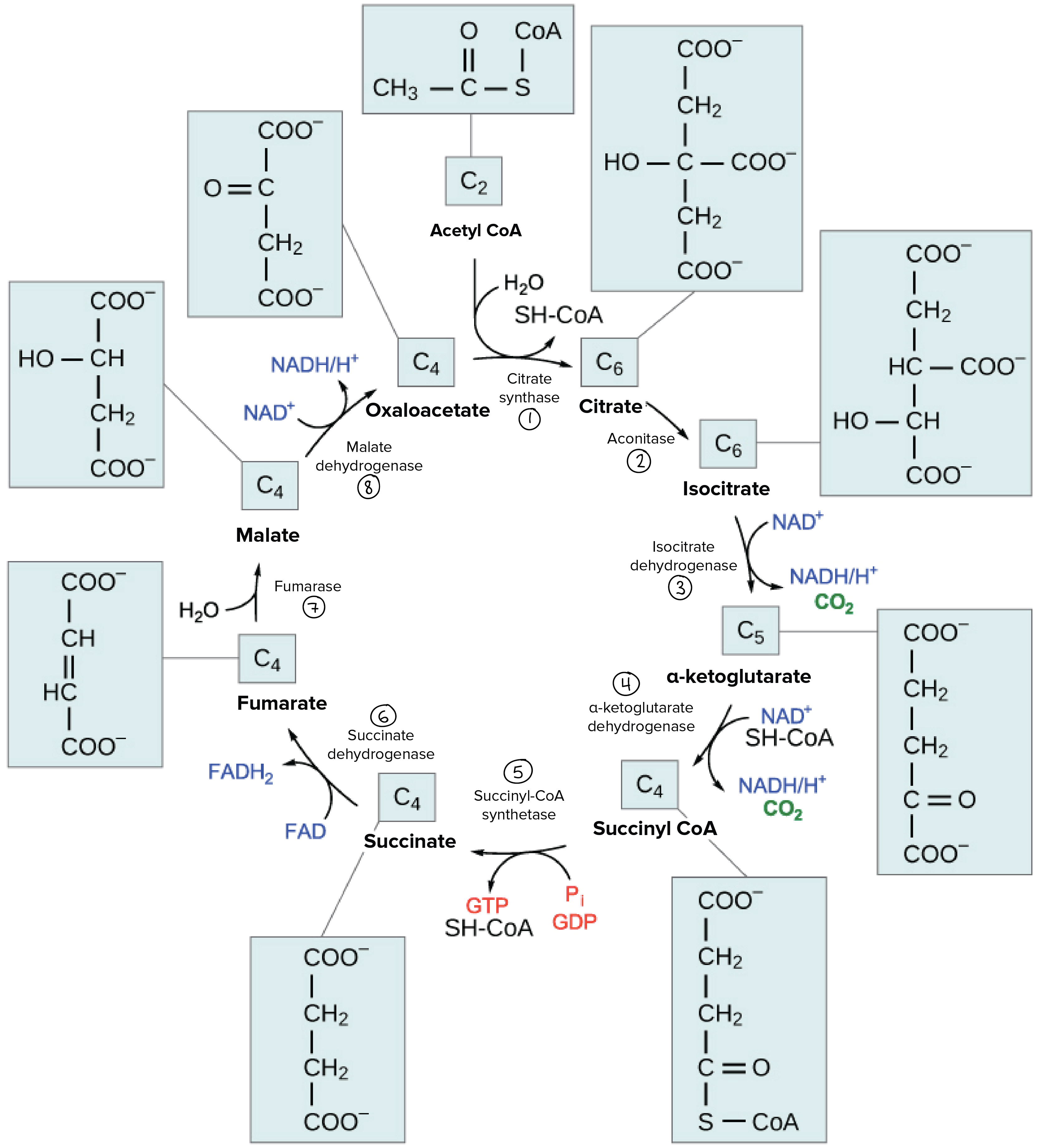
- As outlined above, the main pathway for EtOH metabolism involves successive oxidation of EtOH to acetaldehyde, then to acetate via alcohol and aldehyde DH, respectively.
- Each of these enzymes requires NAD+ and generates NADH in the process.
- As you may recall, NAD+ is a super important electron acceptor for a bunch of processes, including glycolysis, the TCA cycle, and gluconeogenesis.
- An increase in the NADH to NAD+ ratio, as occurs with EtOH ingestion, will therefore have the following effects (a map of glycolysis / TCA cycle / GNG may help contextualize these, but again it’s just for gestalt):
- Most importantly from the perspective of damage to the liver, excess NADH favors the production of glycerol-3-phosphate from DHAP (detail and context provided below). Why does that matter? Because glycerol = backbone for triglycerides! That is, excess EtOH (>60g/d) will favor the esterification of free fatty acids with glycerol to form triglycerides, which will deposit in the liver and contribute to hepatic steatosis, AKA “fatty liver”, the first sign of alcohol-induced liver damage.
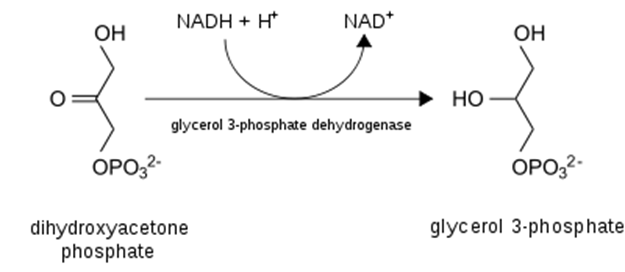

- Necessitate regeneration of NAD+ for ongoing glycolysis by some other means, which, you guessed it, requires generation of lactate from pyruvate — just like in anaerobic conditions in which NADH cannot be used by the electron transport chain (due to lack of O2 as final electron acceptor)
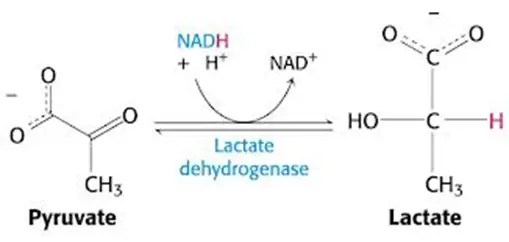
- In other words, you’ll have a lactic acidosis.
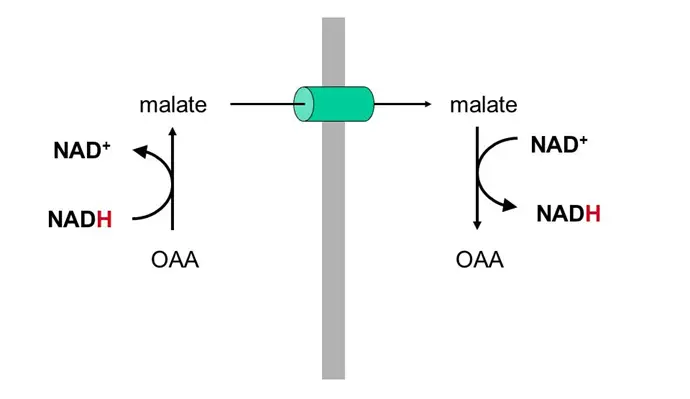
- Favor production of malate from OAA (see below) in the TCA cycle, which sucks because OAA is the starting substrate for gluconeogenesis (OAA –> phosphoenolpyruvate –> glycolysis backwards) — this is why you get the munchies when you drink!!! Your GNG is toast and you’re therefore prone to hypoglycemia!
- Acute EtOH use also increases release of stress hormones including glucocorticoids, which favors the release of free fatty acids from adipose tissue as a way of mobilizing energy stores; these free FAs may then go back to the liver and be packaged as TGs, further contributing to steatosis.
- For some reason, chronic EtOH use also inhibits fatty acid oxidation within the liver and the release of VLDL release from the liver
- Can’t do normal FA oxidation? FAs just sit there and make a metabolic mess (hand-waving, sue me)
- Recall that VLDL carries TGs from the liver to the tissues, and becomes LDL once several FFAs are released (with the help of lipoprotein lipase). LDL then delivers cholesterol remaining in the lipoprotein to the tissues.
- So, if you can’t release VLDL from the liver, the TGs & cholesterol just stay there and cause…steatosis.
- In terms of what causes alcoholic FLD to progress from just steatosis to steatohepatitis (fat + inflammation) to fibrosis (scarring) and then cirrhosis, it seems that
- ***TAKE HOME POINT/tl;dr***: EtOH –> fatty liver.
- **supplement with FA
- For some reason, chronic EtOH use also inhibits fatty acid oxidation within the liver and the release of VLDL release from the liver
- Histopathology
- Epidemiology / Clinical Presentation
- Workup: labs, imaging, other studies
- 2:1 AST:ALT
- Macrocytosis vs megaloblastic anemia
- Thrombocytopenia
- Alk phos/GGT
- Management
- Maddrey/steroids
- Stopping the EtOH
NAFLD/NASH
- Pathogenesis / Etiology / Pathophysiology
- Okay so from the above, it should be apparent that chronic excessive EtOH use can cause fatty change in the liver.
- Increasingly however, with the rise of metabolic syndrome (dyslipidemia + HTN + central obesity + hyperglycemia/glucose intolerance), the most common cause of fatty liver in the USA is no longer alcoholic
- Histopathology
- Epidemiology / Clinical Presentation
- Workup: labs, imaging, other studies
- Management
Viral Hepatitis
WOW WHAT A TOPIC!!!! Let’s start with a summary chart, because there’s a lot to know:
| Virus | HAV | HBV | HCV | HDV | HEV |
| Family | Picornavirus | Hepadnavirus | Flavivirus | Deltavirus | Hepevirus |
| Genome | ssRNA+ | Partially dsDNA | ssRNA+ | ssRNA- | ssRNA+ |
| Acute Mortality | 0.2-0.4% | 1.0-1.2% | 0.2% | 2-20% | 0.2% (20% if pregnant) |
| Chronicity | None | ~10% | ~85% | 2-70% | None (except solid organ transplants) |
| Extra-Hepatic Manifestations | Membranous GN > MPGN Polyarteritis nodosa | MPGN Cryoglobulinemia Lichen planus Porphyria cutanea tarda | |||
| Route of Transmission | Fecal-oral Anal sex | Parenteral Sexual Perinatal | Parenteral Sexual Perinatal | Parenteral Sexual | Fecal-oral Anal sex |
| Clinical Markers | HAV RNA | sAg eAg DNA | HCV RNA | HDV RNA | HEV RNA |
| Antibodies | Anti-HAV Ab (IgM / IgG) | Anti-HBs Anti-HBc (IgM / IgG) Anti-HBe | Anti-HCV Ab | Anti-HDV | Anti-HEV (IgM / IgG) |
**make sure to discuss extra-hepatic manifestations of HCV
Autoimmune Hepatitis
- Pathogenesis / Etiology / Pathophysiology
- Histopathology
- Epidemiology / Clinical Presentation
- Workup: labs, imaging, other studies
- Management
Drug-Induced Liver Injury
- Pathogenesis / Etiology / Pathophysiology
- Acetaminophen / APAP
- Histopathology
- Epidemiology / Clinical Presentation
- Workup: labs, imaging, other studies
- AST/ALT often in thousands
- Management
Hereditary Hemochromatosis
- Pathogenesis / Etiology / Pathophysiology
- Histopathology
- Epidemiology / Clinical Presentation
- Workup: labs, imaging, other studies
- Management
Wilson’s Disease (Hepatolenticular Degeneration)
- Pathogenesis / Etiology / Pathophysiology
- Histopathology
- Epidemiology / Clinical Presentation
- Workup: labs, imaging, other studies
- Management
Alpha-1 Antitrypsin Deficiency
- Pathogenesis / Etiology / Pathophysiology
- Histopathology
- Epidemiology / Clinical Presentation
- Emphysema (esp in non-smoker) + cirrhosis = done
- Workup: labs, imaging, other studies
- Management
Primary Sclerosing Cholangitis
- Pathogenesis / Etiology / Pathophysiology
- Histopathology
- Epidemiology / Clinical Presentation
- Workup: labs, imaging, other studies
- Management
Primary Biliary Cirrhosis
- Pathogenesis / Etiology / Pathophysiology
- Histopathology
- Epidemiology / Clinical Presentation
- Middle aged woman who’s yellow and itchy
- Workup: labs, imaging, other studies
- Management
Jaundice
Biliary Fun:
o Cholelithiasis – lith means stone
o Biliary colic
o Cholecystitis
o Choledocholithiasis
o Ascending cholangitis
· Types of gallstones
o Cholesterol
o Pigmented
Esophageal
Esophagitis – reflux, eosinophilic, infectious
Gastric
Acute Gastritis
Chronic Gastritis
Small + Large Intestine
Malabsoprtive disease (Whipple, CF, Celiac)
Crohn’s Disease
Ulcerative Colitis
Colon Polyps
Colon Cancer
Diverticulosis / Diverticulitis
Pancreatic
Acute Pancreatitis
Chronic Pancreatitis
Pancreatic Neoplasms
Click and check out these popular articles for more information: 🙂
Ectoderm vs Endoderm vs Mesoderm
Circulatory System: Blood Flow Pathway Through the Heart
Circulatory System: Heart Structures and Functions
Ductus Arteriosus Vs Ductus Venosus Vs Foramen Ovale: Fetal Heart Circulation
Cardiac Arrhythmias: Definition, Types, Symptoms, and Prevention
Upper Vs Lower Respiratory System: Upper vs Lower Respiratory Tract Infections
Seven General Functions of the Respiratory System
Digestive System Anatomy: Diagram, Organs, Structures, and Functions
Kidney Embryology & Development: Easy Lesson
Psychology 101: Crowd Psychology and The Theory of Gustave Le Bon
Introduction to Evolution: Charles Darwin and Alfred Russel Wallace
Copyright © 2022 Moosmosis Organization: All Rights Reserved
All rights reserved. This essay or any portion thereof may not be reproduced or used in any manner whatsoever
without the express written permission of the publisher.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Amazing article on gastrointestinal pathophysiology!
LikeLiked by 2 people
Thank you!
LikeLike
Wow, excellent educational guide on the GI system! Very comprehensive. 🙂
LikeLiked by 1 person
Thank you! 😄
LikeLike
Fantastic!
LikeLiked by 1 person
Thanks!
LikeLike